gateTree Demonstration
Published:
A user-informed clustering tree algorithm for cell population identification in flow cytometry data.
Install gateTree.
remotes::install_github("UltanPDoherty/gateTree")
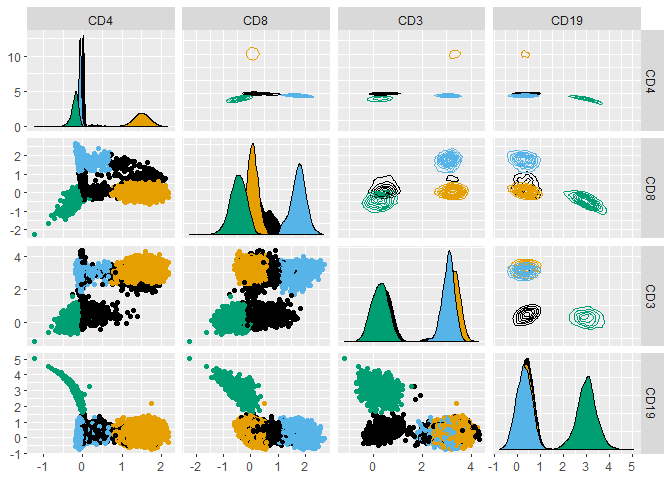
Load and plot data from the healthyFlowData package.
library(healthyFlowData)
data(hd)
hfd1 <- hd.flowSet[[1]]@exprs
GGally::ggpairs(hfd1, upper = list(continuous = "density"), progress = FALSE)
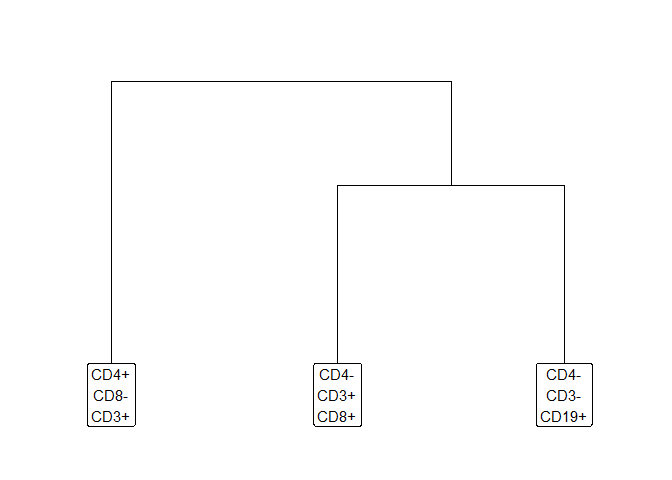
Prepare a plusminus table which describes three populations.
- CD4+ T Cells (CD4+CD8-CD3+CD19-)
- CD8+ T Cells (CD4-CD8+CD3+CD19-)
- B Cells (CD4-CD8-CD3-CD19+)
plusminus1 <- as.data.frame(rbind(
"CD4+_T" = c(+1, -1, +1, -1),
"CD8+_T" = c(-1, +1, +1, -1),
"B" = c(-1, -1, -1, +1)
))
colnames(plusminus1) <- colnames(hfd1)
plusminus1
## CD4 CD8 CD3 CD19
## CD4+_T 1 -1 1 -1
## CD8+_T -1 1 1 -1
## B -1 -1 -1 1
Excel can be used to save or create tables (openxlsx package).
openxlsx::write.xlsx(
plusminus1,
"~/plusminus.xlsx",
rowNames = TRUE,
colNames = TRUE
)
plusminus2 <- openxlsx::read.xlsx(
"~/plusminus.xlsx",
rowNames = TRUE,
colNames = TRUE
)
Run the gatetree function.
hfd1_gatetree <- gateTree::gatetree(hfd1, plusminus2,
min_scaled_bic_diff = 50,
min_depth = 10,
show_plot = c(TRUE, FALSE)
)
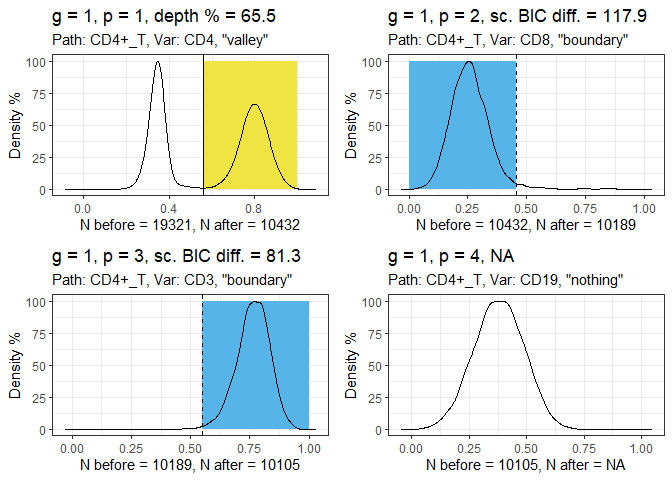
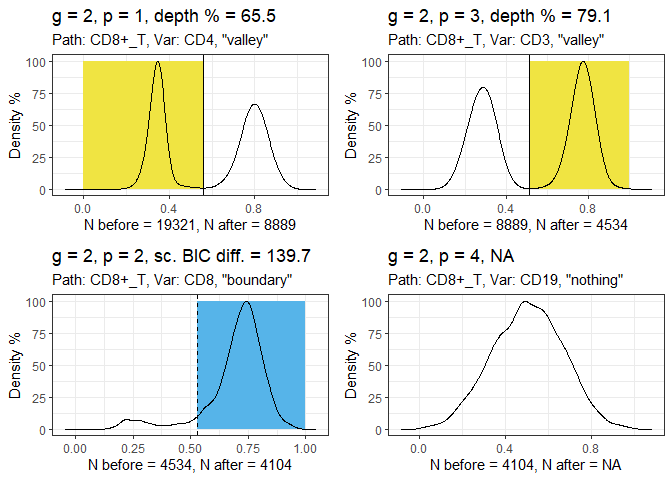
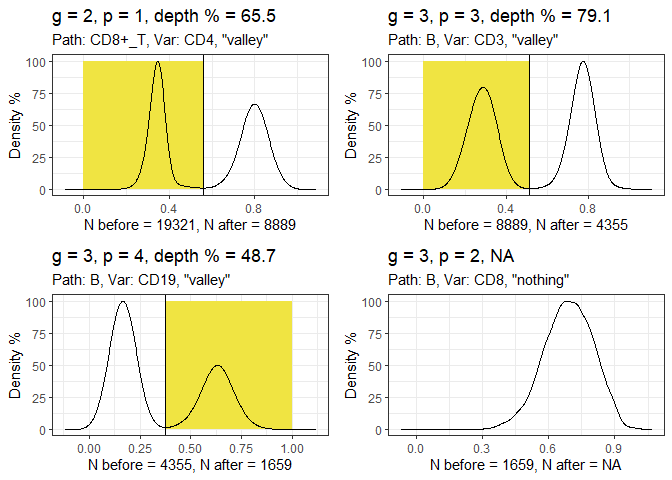
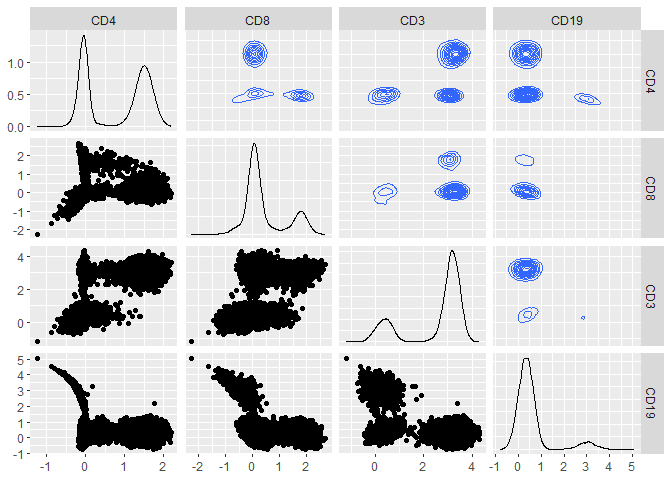
Plot the tree diagram.
hfd1_gatetree$tree_plot +
ggplot2::scale_y_continuous(expand = c(0.1, 0.1)) +
ggplot2::scale_x_continuous(expand = c(0.1, 0.1))
Plot the data, coloured according to the gateTree labels.
GGally::ggpairs(hfd1,
progress = FALSE,
upper = list(continuous = "density"),
ggplot2::aes(colour = as.factor(1 + hfd1_gatetree$labels))
) +
ggokabeito::scale_colour_okabe_ito(order = c(9, 1, 2, 3)) +
ggokabeito::scale_fill_okabe_ito(order = c(9, 1, 2, 3))